
Commons 3 Light 23.50 inch Post Light & Accessory - trans globe lighting
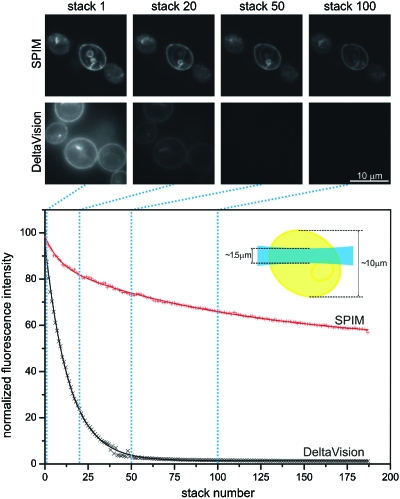
The yeast cells stably expressed Ady2-myeGFP. A stack of 46 planes was acquired every 8 sec. A total of 190 stacks were recorded with a SPIM using a Carl Zeiss Achroplan 100×∕1.0 W lens and an Orca ER CCD camera. The imaging conditions on the two microscopes were adapted to provide comparable signal to noise ratios at comparable excitation intensities. The excitation wavelength was 488 nm while the fluorescence emission was recorded above 510 nm. The calculation of the fluorescence intensity (crosses in the graph) was performed in the central plane of a yeast cell. The measurements were fitted with a double exponential decay function (solid lines). The fluorescence decay in the widefield microscope was approximately six higher than in the SPIM. This number is supported by the fact that only one-sixth of the whole yeast cell is illuminated by the light sheet in SPIM (see the graph inlet) while the widefield microscope illuminates the whole cell for every image recorded. The ratio in bleaching rates is, therefore, even bigger for larger specimens. It should be stressed that the imaging conditions in such experiments will never be perfect since the sample preparation conditions and samples themselves tend to vary naturally. The yeast cells were obtained and imaged in collaboration with Christof Taxis and Michael Knop.
U.K. received support from the Louis Jeantet Foundation. E.G.R. and E.H.K.S. acknowledge the support from the German Ministry of Research (BMBF—Project QuantPro). We would like to thank Christof Taxis and Michael Knop for the yeast samples as well as Petra Jakob for the Cytodex sample.
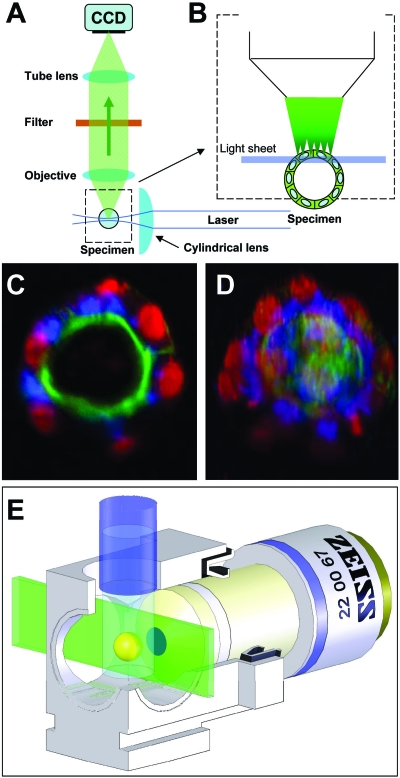
The detection unit is a simplified fluorescence wide-field microscope. An objective lens, a filter, and a tube lens form the fluorescence image on the wide-field detector (e.g., a CCD camera). The objective lens defines the focal plane. The illumination unit generates the light sheet for the illumination of the volume around the focal plane in the specimen. The movement unit holds the specimen and moves it relative to the optical setup, which is usually at rest. Three translation and one rotation stage position scan the specimen. The specimen can be immersed in a medium-filled chamber for optimal experimental conditions. Finally, the control unit, a standard computer equipped with data acquisition boards, controls the hardware and acquires the data. Additional optical elements such as beam couplers and splitters for auxiliary units can be introduced in the infinity corrected space (ICS) between the objective lens and the tube lens and will require further lasers and other optical components not shown here (Nanotool port) (m: mirror).
In confocal and conventional fluorescence microscopy, the illumination and detection axes use the same objective in an antiparallel manner commonly known as epifluorescence. Since the lateral extent of the point spread function (PSF) is smaller than its axial extent, the use of a single lens always results in an anisotropic PSF, which is elongated along the z axis. In LSFM, the specimen is illuminated from the side by a thin light sheet. The center of the light sheet overlaps with the focal plane of the detection system. Hence, the emitted light is detected along a detection axis, which is orthogonal to the illumination axis (Fig. 2). The LSFM should be regarded as a combination of at least two independently operated illumination and detection systems. The resolution of an LSFM is defined by the properties of the detection and the illumination setups. The lateral resolution is only determined by the detection objective lens and is the same as in a regular fluorescence wide-field microscope. When operating at a numerical aperture (NA) below 0.6, the axial resolution is dominated by the thickness of the light sheet and is less affected by the properties of the detection lens. The situation is, thus, different than in an epifluorescence setup. Therefore, contrary to other microscopes, the isotropy of the PSF is much better at low NAs (Engelbrecht and Stelzer 2006). However, even for high NA systems, the observed volumes are in the same range as those of confocal fluorescence microscopes.
LED Backlight strip 6 lamp For HRS_SQY39GA064_3X6_2W_MCPCB 39D3503V2W6C1B73112M NVR-8060-39RD2S-SMA-N NVR-7707-39RDS2 3V/led ... LED lamps, ensuring bright and ...
There are at least three reasons that make LSFM well suited for this kind of multiple-views imaging: (i) specimens are not attached to flat surfaces and can be rotated by a full angle, (ii) long working distance lenses are usually used that provide enough space for a comfortable rotation of big specimens (up to several mm), and (iii) images produced by LSFM are well suited for further image processing due to their low noise and high dynamic range.
In LSFM, the specimen is no longer positioned on a microscope slide, but placed in a liquid filled specimen chamber. Four main specimen preparation techniques have been developed and used to perform LSFM: hooking, embedding, containing, or flat mounting (Fig. 6).
The resolution of LSFM can be further improved by using its tomographic capability. In fact, the resolution within the focal plane (i.e., lateral resolution) and the resolution along the optical axis of any microscope are determined by the numerical aperture of the objective lens. The axial extent of the PSF is, however, at least three times larger than the lateral extent of the PSF. The combination of multiple views of the same volume along different directions can result in a single three-dimensional data set with an isotropic resolution that is dominated by the lateral resolution of the detection system (Swoger et al. 2003, Verveer et al. 2007, Swoger et al. 2007) (see Digital image processing of LSFM images below).
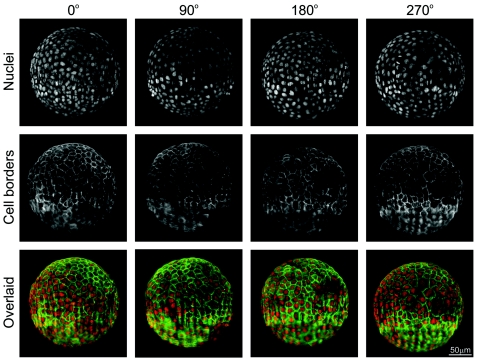
MDCK cells that stably express an E-cadherin EGFP construct were grown on a Cytodex 3 microcarrier (mean diam. 200 μm, GE Healthcare) for two weeks prior to imaging with the SPIM. The nuclei were stained with Draq5 (Biostatus), the cell borders and, thus, the locations of the cells’ outer plasma membrane are highlighted by the E-cadherin EGFP expression. The Cytodex bead was fixed in paraformaldehyde and then embedded in 1% agarose in a capillary (inner diam. 1,1 mm) It was imaged using a 20× objective (Zeiss Achroplan; NA 0.5) along four angles (0, 90, 180, and 270 deg). Each three-dimensional stack of images consists of 1,033 images (z spacing, 0,32 μm). The total multiple-views super set of images represents 4,132 images (i.e., 1,1 GB).
Finally, the control unit operates the hardware and controls the data acquisition process. The control unit manages the interaction between the different units during the data recording process. The computer also provides the user interface and takes care of data handling issues.
In summary, the method of specimen preparation for LSFM must respect three very important criteria. It must be mechanically stable. The specimen must be well supported to avoid distortion due to movement during imaging. It must support the physiological aspects of the specimen (e.g., development). The mounting system should be flexible enough to allow the specimen to develop and should not change its mechanical properties during observation or dissolve in the buffer. Finally, it must support good imaging (e.g., no mechanical interference during the imaging). For example, the refractive index of the mounting medium should be as close as possible to the refractive index of the buffer filling the imaging chamber in order not to scatter light. Both optical and physiological aspects have to be brought together, especially in live imaging where biocompatibility has to be considered.
The second and most common technique is embedding. The specimen is embedded in a gelling agent, usually low melting agarose, using a specimen holder such as a glass capillary that shapes a cylinder [Fig. 6B]. This agarose cylinder is pushed out of the capillary and placed in front of the objective lens. This provides a complete access to the specimen. The chosen gelling agent has a refractive index, which is close to that of water and strong enough not to break during translation or rotation. This specimen preparation technique has been extensively used for imagingD. melanogaster (embryo, larvae, pupa, and adult) (Swoger et al., 2007), A. gambiae, fixed cysts and cell aggregates (Pampaloni et al., 2007), C. elegans, D. rerio (Huisken et al., 2004; Swoger et al., 2007), O. latypes, yeast cells (Taxis et al., 2006), or zooplankton. One limit of embedding is the effects of the gelling agent on the specimen. It may exert compression forces on the specimen during the gelling process, especially at high concentrations (e.g., 2% agarose). It can also restrain the specimen movements and this can be crucial when imaging developing samples such as embryos.
The optical resolution within the focal plane (lateral resolution) and the resolution along the optical axis of any microscope are determined by the numerical aperture of the objective lens. The axial resolution is, for basic physical reasons, always multiple times worse than the lateral resolution. No deconvolution algorithm can bridge this gap and improve an image produced by a standard single lens microscope to have an isotropic resolution.
In a LSFM, a CCD camera is used to collect a two-dimensional image. Modern CCD chips provide a high quantum efficiency (up to 70% for standard CCD, up to 95% for back-illuminated electron-multiplying CCD), while laser-scanning microscopes rely on photomultiplier tubes with quantum efficiencies up to 30% and usually below 20%. More importantly, since a laser-scanning microscope is a sampling device and measures one pixel at a time, it has to divide the total time needed to acquire an image among all pixels in an image. For a standard-sized image (512×512 pixels), this means that the microscope spends only about 1–10 μsec measuring the fluorescence intensity for every pixel at a standard frame-rate (1–5 frames per sec). A CDD chip, on the other hand, collects the image for all pixels of a two-dimensional grid “in parallel.” This allows much longer per-pixel measurement times at identical frame rates, which in turn means that more fluorescence photons can be collected. While in a standard confocal fluorescence image the brightest pixels correspond to only 10–100 detected photons, CCD chips can collect up to several thousand photons from those same pixels at an even higher frame rate. This gives us an estimate of the dynamic range of such images: 3–5 bits with confocal fluorescence microscopes compared to 12–14 bits in LSFM.
Like in any other microscopy technique, the specimen preparation must be carefully considered, as badly labeled and deformed specimens will not take full advantages of the microscope. This is especially important in LSFM as the specimen handling is performed in a very different way (rotation, incubation chamber . . .). In general, however, the attitude should be that LSFM provide an entirely new means for specimen preparation. The fact that illumination and detection occur along different directions should be regarded as an opportunity to reconsider the means according to which specimens can be prepared. After all, we want to prepare specimens in a manner that maintains it close to its actual physiological appearance.
LEDilluminationlights
Until now, confocal and two-photon fluorescence microscopies provided the most popular solutions to image relatively thick specimens with a reasonable resolution and a moderate penetration depth (Fig. 1). In confocal fluorescence and in wide field fluorescence microscopy, the illumination light excites fluorophores along the entire thickness of the specimen. In confocal fluorescence microscopy, the out-of-focus light is discriminated by the pinhole in front of the detector. So, most of the light is not detected but its damaging effects (in particular photo toxicity and photo bleaching) will affect the entire specimen even if only one plane is imaged at a time. Another issue in confocal fluorescence microscopy is its limited penetration depth, especially with high numerical aperture objective lenses. An approach to improve the penetration depth seems to be multiphoton fluorescence microscopy. Its penetration depth is probably 1.5–2× times higher than that of confocal fluorescence microscopy. However, its lateral resolution is worse than that of any conventional fluorescence microscope (Stelzer et al., 1994) and it requires considerably higher light intensities than any other technique. Its use, thus, leads to significant photo toxic and photo bleaching effects.
We are known in the industry for offering quality Star Ceiling Fibre Optic Lights. In this range, we also offer luxury crystal chandeliers that are made using ...
Infrared radiation is emitted by any object that has a temperature (ie radiates heat). So, basically all celestial objects emit some infrared. The wavelength at ...
Techniques such as optical projection tomography (Sharpe et al., 2002) have been developed to image large specimens such as complete organs or developing embryos (Fig. 1). Since this technique observes a specimen along multiple directions, one obtains a three-dimensional reconstruction and a better access to the three-dimensional fluorophore distribution. A specimen can be imaged along different directions and the different views are fused into a single volume.
The illumination unit generates the light sheet that is used to excite the fluorophores. It overlaps with the focal plane in the specimen. The laser provides a collimated beam. An acoustooptic tunable filter (AOTF) picks at least one of several laser wavelengths and adjusts its intensity. A beam expander changes the diameter of the collimated beam and feeds it into an optical device that creates a light sheet. A very simple setup would use a single cylindrical lens. However, cylindrical lenses with the required NA are not easily available as well-corrected elements and tend to introduce aberrations. Therefore, an objective lens is used in combination with a cylindrical lens that is rotated by 90 deg (Greger et al., 2007) (Fig. 5).
Accentlighting
by Y Chen · Cited by 2 — We introduce a cost-effective, scalable, and versatile high-resolution imaging framework, called projected Light Sheet Microscopy (pLSM),
Images produced by LSFM have a higher dynamic range and a better signal-to-noise ratio than those produced by confocal fluorescence microscopes and are, therefore, better suited for digital image processing.
Official websites use .gov A .gov website belongs to an official government organization in the United States.
20201221 — An international collaboration of researchers has cast doubt on this principle by measuring non-zero transverse spin values in totally unpolarized light.
The use of light sheet in optics was introduced by Heinrich Siedentopf in 1903 with the development of the slit ultramicroscope together with Richard Zsigmondy (Siedentopf and Zsigmondy, 1903). Although it has been suggested as an imaging tool in biology several times (Voie et al. 1993; Huber et al., 2001; Fuchs et al., 2002), its use for fluorescence microscopy was not realized until a few years ago (Huisken et al., 2004). The theory has been mainly described by Stelzer and Lindek (1994) in their descriptions of various different confocal theta fluorescence microscopes. The development of different methods has been mainly driven by the requirements imposed by the specimen (insect, marine bacteria, fish embryo, or mouse brain). Most of the LSFMs have a horizontal design, i.e., the illumination and the detection axis are oriented horizontally. The ultramicroscope (a replica of the original Siedentopf-Zsigmondy design) is based on a vertical design; the detection axis is vertical and uses a two-sided light sheet illumination (Dodt et al., 2007). This influences the specimen manipulation, especially if we consider rotation.
The same holds true in confocal fluorescence microscopy. Optical sectioning is not obtained in the illumination process, but by discriminating against the out of focus fluorescence light with a pinhole in front of the detector. Hence, most of the light emitted by the specimen does not reach the detector. Using high NA lenses, about 80% of the emitted light remains undetected. Moreover, the image is obtained by sampling the specimen one point at a time. Different planes are acquired by scanning the sample at different z positions to obtain the three-dimensional fluorophore distribution. This results in a much higher exposure of the sample (essentially by a factor determined by the number of the acquired planes). An LSFM, which acquires a stack of images, illuminates each plane only once. The fluorophores outside the focal plane are not illuminated and, therefore, not subject to photo-damage. Compared to confocal and conventional fluorescence microscopy, the fluorophores are used very efficiently and photo damage (e.g., photo bleaching) is greatly reduced (Fig. 3). The improvement can be quantified by the ratio of the light sheet thickness over the specimen thickness. In fact, if one uses a 1.5 μm light sheet to observe a 10 μm thick yeast cell, one would need six planes to scan the entire volume. In LSFM one illuminates every plane once for each stack. In confocal fluorescence microscopy, one illuminates the entire volume each time one acquires a plane. In our yeast example, the LSFM will reduce the energy load on a specimen by a factor of six (Fig. 3). This factor increases with the size of the specimen (20–30 for a single cell, up to 500 for an entire embryo). Hence, LSFM photo-toxic effects are greatly reduced compared to other microscopic techniques. This improvement can be used to significantly increase the period of observation, the speed of imaging, the number of views per given volume, or in many other ways that seem suitable to the experimenter.
The LSFM follows a very open concept and is, thus, a very flexible microscope. The implementation of other fluorescence techniques is straightforward. The infinity corrected space (ICS) in the detection unit can be used to couple in or out additional devices, e.g., laser microsurgery (Engelbrecht et al., 2007).
Light-sheet-based fluorescence microscopy (LSFM) is a fluorescence technique that combines optical sectioning, the key capability of confocal and two-photon fluorescence microscopes with multiple-view imaging, which is used in optical tomography. In contrast to conventional wide-field and confocal fluorescence microscopes, a light sheet illuminates only the focal plane of the detection objective lens from the side. Excitation is, thus, restricted to the fluorophores in the volume near the focal plane. This provides optical sectioning and allows the use of regular cameras in the detection process. Compared to confocal fluorescence microscopy, LSFM reduces photo bleaching and photo toxicity by up to three orders of magnitude. In LSFM, the specimen is embedded in a transparent block of hydrogel and positioned relative to the stationary light sheet using precise motorized translation and rotation stages. This feature is used to image any plane in a specimen. Additionally, multiple views obtained along different angles can be combined into a single data set with an improved resolution. LSFMs are very well suited for imaging large live specimens over long periods of time. However, they also perform well with very small specimens such as single yeast cells. This perspective introduces the principles of LSFM, explains the challenges of specimen preparation, and introduces the basics of a microscopy that takes advantage of multiple views.
An LSFM consists of four basic units, which address (1) illumination of the specimen (light sheet formation), (2) translation and rotation of the specimen, (3) light detection, and finally (4) control of different mechanical and electronic parts, collection, and postprocessing of the data (Fig. 5).
Even if one is only interested in the information in the focal plane, a conventional fluorescence microscope illuminates the entire specimen for every plane that is imaged.
Illumination lightingmeaning
(A) The simplest method is hooking or clipping the specimen in front of the objective. (B) The specimen can be embedded into a cylinder of gelling agent such as low melting agarose. (C) The specimen can be contained into a chamber made of agarose or a transparent polymer. (D) Finally, the specimen can be prepared and fixed on a coverslip and imaged at an angle in regard to the light sheet.
LED is the modern, environmentally friendly, cost effective way to light fibre optics and our light sources all feature industry standard 30mm fibre ports, so ...
The number of collected photons has a great impact on the noise level of the image. The emission of a photon by a fluorophore and its subsequent detection is a single random event and, therefore, follows Poisson statistics. The average rate at which photons are emitted from a small volume is proportional to the number of fluorophores in that volume and can be accurately measured only if a sufficient number of photons is collected. The error in the fluorophore density measurement is proportional to the square root of the number of detected photons. The signal to noise ratio, therefore, equals the square root of the number of detected photons and is usually 3–10 in the case of laser-scanning fluorescence microscopes and in the range of 50–200 for LSFM. The image is usually further degraded by other sources of noise, such as dark current, electron multiplication, read out, and laser intensity fluctuations. They are normally less important than the Poisson noise. Their relative magnitude is, in general, also reduced with an increasing number of detected photons, further increasing the gap between signal-to-noise ratios of fluorescence laser scanning and LSFM microscopes.
Once a set of multiple-views images is recorded, it is digitally fused into a single image. This process can be split into three parts. During the initial preprocessing, all information about the images is used to force all single views into the same orientation. The physical rotation of the specimen in the microscope is, thus, digitally reversed. Next, all single views are aligned relative to each other with subpixel precision. Considering the vast size of the LSFM image stacks and the storage space of computers, it is more convenient to use simple image registration transformations, e.g., translation only. This, of course, requires extremely precise image acquisition and preprocessing, so that the preprocessed images fit without further scaling and rotation.
This stereo microscope fiber optic cold light source provides brightness higher than 150 W halogen light source yet consumes 80% less energy.
IM Vellekoop · 2007 · 2103 — We demonstrate focusing of coherent light through disordered scattering media by the construction of wavefronts that invert diffusion of light. Our method ...
Precision methods are, e.g., stimulated emission depletion (STED) (Willig et al., 2006), photoactivated localization microscopy (PALM) (Betzig et al., 2006), and stochastic optical reconstruction microscopy (STORM) (Rust et al., 2006). MRI—magnetic resonance imaging, PET—positron emission tomography, OPT—optical projection tomography (Sharpe et al., 2002), OCT—optical coherence tomography, LSFM—light-sheet-based fluorescence microscopy, dmvLSFM—deconvolved multiple views light-sheet-based fluorescence microscopy. Figure inspired by Roger Tsien (Tsien 2003).
As mentioned above, LSFM images are well suited for further deconvolution due to their excellent signal-to-noise ratio and their high dynamic range (Verveer et al., 2007; Swoger et al., 2007). Furthermore, since objective lenses with long working distances and consequently smaller numerical apertures are often used in LSFM, deconvolution can greatly improve the visual impression of resulting images. Deconvolution does not improve the resolution.
Types ofilluminationPDF
Life is all about dynamic processes of complex multicellular organisms in a three-dimensional world. To analyze, describe, and understand life requires us to observe dynamic three-dimensional processes with a sufficient spatiotemporal resolution and in great detail. However, high specificity is currently achieved with fluorescence labeling, which has the disadvantage that it interferes with the specimen and provides a relatively weak signal. In addition, fluorophores are “consumed” during the excitation process. Fluorescence imaging has, thus, remained a challenge for the last 30 years. The ideal fluorescence light microscope should provide images of three-dimensional fluorophore distributions inside a specimen with high resolution, without morphological distortions, within short time intervals, and over extended periods of time. It should allow scientists to perform long-term time lapse imaging without losing the fluorescence signal and with a limited stress for the live specimen. So, how close have we come? How fast and how long can we image without losing the signal? Which resolution can be achieved?
The detection unit is essentially a fluorescence wide-field microscope. It consists of an objective lens, a filter wheel, a tube lens, and a wide-field detector (e.g., a CCD camera). Since LSFMs deal with biological specimens, they mainly rely on water dipping lenses, but they can also use standard air lenses for lower magnifications. A single emission filter blocks the excitation light and transmits the fluorescence signal emitted by the specimen. A dichroic mirror is not required. The primary image is formed on the camera via a tube lens. The actual type of the camera depends on the application and will vary in the number of pixels and lines, the dynamic range, and the frame rate. An LSFM can include a Nanotools port (Greger et al., 2007; Engelbrecht et al., 2007) within the infinity corrected space of the detection unit (Fig. 5). It allows the implementation of various optical manipulation techniques: laser nanosurgery (UV laser), optical tweezers (IR laser), fluorescence recovery after photobleaching (FRAP), and photoactivation of GFPs (paGFP).
UV Flashlight 51 LED 395 Nm Ultraviolet Blacklight Pet Urine Detector Dry Stains and Bed Bugs Detector. 4.6 ( 44 reviews ).
(A) The central element of an LSFM is a regular fluorescence microscope. It consists of an objective lens, a filter, a tube lens, and a wide-field detector. The specimen is illuminated from the side by focusing a collimated laser beam to a light sheet with a cylindrical lens. (B) In the specimen, only those fluorophores that are actually observed are also illuminated. This is equivalent to a true optical sectioning but does not generate photo damage outside the focal plane. (C) A single image out of a stack of 120 planes, which was recorded inside a fixed MDCK cyst, demonstrates the optical sectioning capability, the excellent signal to noise ratio, and the extremely low background. (Red: DRAQ5 staining of the nuclei; blue: GM130 antibody staining of the Golgi apparatus; green: Phalloidin-Alexa488 of the actin network). (D) Projection of the stack of 120 images recorded at different depths. (E) A picture of a setup showing the incubation chamber and the specimen during imaging.
Laser-based microsurgery is a versatile tool with an increasing number of applications in biological research from microtubule dynamics (Colombelli et al., 2005), actin dynamics (Colombelli et al., 2006), and developmental or morphogenetic studies (Grill et al., 2001). However, laser microsurgery has often been restricted to two-dimensional cultures because of the difficulty in imaging living 3D specimens and applying an appropriate 3D-shaped ablation pattern. Our laser microsurgery instrument was integrated into a LSFM by coupling the ablation beam into the microscope’s detection path using a dichroic mirror. This setup provides three-dimensional specimen ablation and quasi-simultaneous acquisition of an optically sectioned three-dimensional fluorescence image (Engelbrecht et al., 2007). It has performed very well over a wide range of scales from dissecting single microtubules to disrupting single cells in spherical cysts and performing cuts greater than 100 μm long in a zebrafish fin. It could also be used to study dynamic processes, i.e., immune cell response in live Drosophila. This 3D cutter represents a very important implementation in LSFM, as it allows physical manipulation in an entire living specimen.
Illumination lightingdesign
In LSFM, the azimuthal optical arrangement is usually fixed. Therefore, the specimen must be moved along the optical axis of the detection system and through the common volume of the detection focal plane and the illumination plane to acquire three-dimensional stacks of images. The three computer-controlled motorized translation stages move the specimen along three dimensions, and one rotary stage rotates the specimen around the vertical axis. This allows us to access any part of the specimen and to acquire stacks of images along different directions. Additional degrees of freedom (nutation and declination) can be introduced to provide more flexibility when accessing more complicated specimens. Most of the LSFM implementations use a medium filled chamber to maintain and image the specimen. The simplest chamber is a cube made of optical glass or with four glass windows that allow the illumination and the detection of the light at 90 deg (Dodt et al., 2007; Greger et al., 2007). A window can be replaced by a rubber sealing ring such that water dipping lenses can be inserted into the chamber. This has the advantage that no glass surfaces and, hence, changes in refractive index cause aberrations in the optical path of the detection system. This is ideal for biological specimens, because the specimen medium and immersion liquid are identical. Moreover, such a chamber system provides an optimal environment for live specimen imaging and different types of chambers can be used depending on the applications (temperature controlled, pressured, CO2, . . .) (Pampaloni et al., 2007).
There are generally two types of lighting that can be used for microscopes: transmitted and reflected illumination.With transmitted lighting, light shines from behind an object (usually a transparent target) to illuminate the features within that target, while reflected light is used to illuminate the surface of an object. Reflected light can be coaxial (brightfield) or from an angle (darkfield). Coaxial lighting travels along the same direction as the optical path of the lens and is used to view flat surfaces. Ring illumination reflects off of the angled surfaces of an object and is usually used for imaging objects with textured or irregular surfaces.
LSFMs have a relatively small number of components, the three most expensive ones being the laser, the stage, and the camera. However, so far all described light-sheet-based instruments are experimental setups developed by various groups (Voie et al., 1993; Huber et al., 2001; Fuchs et al., 2002; Huisken et al., 2004; Greger et al., 2007; Dodt et al., 2007; Huisken and Stainier, 2007).
Lighting · Automotive Lighting. Flood Lights & Spot Lights. 32 Items. All ... 27.99. Add to Cart Add to List. 6 in. LED Spot Light. ROADSHOCK. 6 in. LED Spot ...
LSFM is a new fluorescence microscopy technique that uses a thin light sheet for the illumination of the focal plane of the detection objective lens. This provides optical sectioning. This microscope combines the advantages of wide-field methods (imaging speed, dynamic range) with those of confocal setups (optical sectioning). For the very first time, it offers a reasonable technique to apply multiple-views imaging in light microscopy. It offers the possibility to obtain quantitative three-dimensional maps of the distribution of multiple fluorophores, for example, the expression pattern of GFP-labeled protein, with high spatiotemporal resolution and an excellent signal to noise ratio. Since LSFM uses the fluorophores orders of magnitude more efficiently than comparable confocal or conventional fluorescence microscopes, LSFM induce much lower photo toxicity and much less photo bleaching. This enables imaging with a higher temporal resolution or over longer periods of time. Mounting of the specimen, e.g., in agarose gels allows the imaging of live specimens under almost physiological conditions. In the coming years, the commercialization of this technology and the increasing numbers of users and developers will open new fields of investigations in cell biology, developmental, and particularly where three-dimensional imaging is most needed: in the stem cell niches and in tissue engineering.
Another recent development of the LSFM is the implementation of structured illumination (SI). Structured illumination was introduced to wide-field fluorescence microscopy as a clever means to discriminate against out-of-focus light (Neil et al., 1997). The structured illumination was combined with a SPIM by lateral modulation of the light sheet intensity (Breuninger et al., 2007). This clearly reduces effects of the scattered light and improves the contrast. The SPIM-SI improves the image quality in strongly scattering specimens.
Types ofilluminationin slit lamp
There are many different approaches to image deconvolution. They can be divided into two groups: nonblind, where the “shape” of the blur is known (e.g., maximum likelihood estimation, Lucy-Richardson algorithm, constrained Tikhonov-Miller algorithm) and blind deconvolution algorithms (e.g., maximum likelihood blind deconvolution, Ayers-Dainty algorithm). A number of different free (e.g., ImageJ plugins) and commercial (e.g., Huygens Deconvolution Software from Scientific Volume Imaging and AutoQuant from MediaCybernetics) computer programs are available today, utilizing both kinds of algorithms.
The first and simplest way consists of “hooking” the specimen using a clip, tweezers, or a hook made of glass, stainless steel, or plastic material [Fig. 5A]. This is particularly suitable to image large specimens such as organs (e.g., mouse brain), adult insects, but also yeast cells dispersed in an agarose droplet, as the hook provides a higher stability of the specimen than holding as a block of agarose (Taxis et al. 2006). However, one has to consider that mechanical contact can partially damage the specimen or interfere with imaging.
In the early 1990s, confocal fluorescence microscopy was further improved with the development of the confocal theta fluorescence microscope (Stelzer and Lindek 1994). It provided a new tool for the investigation of large specimens with a high and isotropic three-dimensional resolution. The fundamental principle was the detection of fluorescence light at a 90 deg angle to the illumination axis. The azimuthal arrangement reduces the out of focus light that has to be discriminated against in the confocal fluorescence microscopy by the pinhole and reduces photo toxicity and photo bleaching. Further research into the theta principle recently resulted in the implementation of the single plane illumination microscope (SPIM), an implementation of LSFM (Huisken et al., 2004; Greger et al., 2007). It is a wide-field version of the confocal theta microscope. The specimen is illuminated with a thin light sheet that results in an intrinsic optical sectioning. Unlike conventional or confocal fluorescence microscopes, which expose the entire specimen for each plane they record, SPIM or LSFM in general only illuminate and observe one plane at a time (Fig. 2).
The illumination axis of most of the implementations uses only one light sheet to illuminate the specimen. However, it is possible to use two light sheets to illuminate the specimen more evenly along two opposite directions both at 90 deg relative to the detection axis (Huisken and Stainier, 2007; Dodt et al., 2007). In order to allow more flexibility for the illumination unit, it is also conceivable to replace the cylindrical lens by a galvanometric mirror to generate a scanned light sheet.
The most straightforward way to improve the axial resolution is based on multiple-views microscopy. The core idea is to observe the same specimen independently along multiple different directions and then use digital image processing techniques to fuse individual images or stacks of images into a single, high quality, three-dimensional image. Such an image can be better than any of the single-view input images in two respects: (i) resolution can be better and more isotropic and (ii) the image of an opaque specimen can be more complete, combining images of parts of the specimen that are only well exposed in some of the single views (Swoger et al., 2003, 2007).
The most commonly used image processing technique is probably filtering. A special case is deconvolution, which takes the properties of the imaging system explicitly into account. The optical resolution of standard microscopes is limited by the wave nature of the light and is determined by the wavelength of the light and the numerical aperture of the objective lens. The limited optical resolution causes blur in the images that can be computationally modified using deconvolution algorithms.
The fourth technique is to mount an object on a flat surface. This is mandatory for the ultramicroscope as the vertical detection axis allows depositing the object underneath the objective lens (Dodt et al., 2007) but in the case of a horizontal detection axis as in the SPIM, the flat surface must be mounted on a holder [Fig. 6D].
So what is the optimal solution for a microscope that addresses the needs of modern biology: a tomographic confocal microscope that combines the advantages of both techniques? Light-sheet-based fluorescence microscopy (LSFM) is indeed a promising answer. We believe that this fluorescence imaging technique addresses crucial issues such as photo bleaching and imaging speed. This perspective covers the principles of this new type of fluorescence microscopy in comparison to other techniques and describes its advantages, its limits, its implementations, and also its challenges including specimen preparation and image processing.
The third mounting technique consists of holding the specimen in a container. This container can be made from a gelling agent using a special moulding device or suitable polymers (refractive index, thickness . . .) [Fig. 6C]. These containers can be held using suitable clips. This technique is convenient to image living cells embedded in three-dimensional extracellular matrices, compression-sensitive specimens (e.g., developing embryos), as well as in vitro assays (Engelbrecht et al., 2007; Keller et al., 2008). The main limitation of this approach is the capacity to tailor the chamber size to the specimen. It is difficult to prepare agarose chambers with an inner diameter of less than 0.5 mm and a beaker wall thin enough not to affect the imaging quality. In addition, suitable polymers are often difficult to find and to shape to accommodate small specimens such as sea urchin eggs (100 μm).
The term tomography should refer to all techniques that produce a single image of a slice or slab (tome in Greek) of a specimen. Confocal fluorescence microscopy and LSFM optically section an object and should be regarded as tomographic techniques. However, this term is currently used for techniques (e.g., CT and OPT) that produce a projection. The image is calculated by performing a back projection of a series of images generated by rotating a specimen around a central axis. In contrast, an LSFM produces a single real image and stacks of real images, which are comprehensible without any further processing. Apart from the fact that LSFM observe along an axis normal to the illumination plane this is of course a major difference between LSFM and e.g., OPT. In conventional and confocal fluorescence microscopes, the specimen is mounted on a flat surface that limits the access to the specimen. In LSFM, the specimen is mounted on a stage that allows movement along all three dimensions and rotation around a vertical axis (see the following paragraph). The sectional images can be recorded along different directions by moving and rotating the specimen (Fig. 4). This makes hidden regions visible and allows researchers to delve deeper into the specimen, e.g., tissue.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
There are different ways to record images along different directions relative to the specimen. While some rely on multiple independent objective lenses pointing towards the same volume (Swoger et al., 2003), others use a single detection lens and rotate the specimen in front of the detection lens. This technique can be easily implemented with an LSFM.



 Ms.Cici
Ms.Cici 
 8618319014500
8618319014500